




LATEST
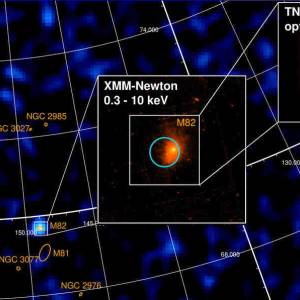
University of Geneva researches a massive magnetic...
Wed, Apr 24th 2024A groundbreaking discovery has been made by the...

Supercomputer simulations uncover the dynamics of...
Tue, Apr 23rd 2024New research conducted by a team of scientists...
AI tool predicts responses to cancer therapy using...
Thu, Apr 18th 2024A recent study conducted by researchers from...

NASA's Near Space Network enables the PACE Climate...
Wed, Apr 17th 2024NASA's PACE mission achieved a significant...
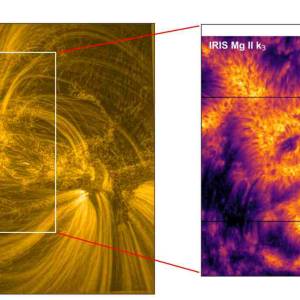
Scientists utilize supercomputer simulations to...
Wed, Apr 17th 2024Cutting-edge research and supercomputer...